

Research Overview
Macrocyclic Peptide Inhibitors of Protein-Protein Interactions
A major research thrust in our laboratory concerns the development of new strategies to direct the synthesis, diversification, and evolution of macrocyclic peptides as potent and selective inhibitors of protein-protein interactions (PPIs). PPIs are implicated in all cellular processes from signal transduction to gene regulation, cell proliferation and apoptosis. Many diseases, including various forms of cancer, arise from or depend upon aberrantly misregulated PPIs. Chemical agents capable of targeting PPIs with high potency and selectivity can provide invaluable tools for the study of cellular pathways and useful starting points for the development of new therapeutics. However, the development of such compounds has represented a major challenge in chemical biology and drug discovery. In the Fasan group we are investigating the potential of macrocyclic peptides for tackling this challenge. Macrocyclic peptides combine a compact (500-2,000 Da) and conformationally semi-rigid architecture with a high degree of functional complexity, which make them attractive scaffolds for selective and high-affinity recognition of a target protein-protein interface and thus for disrupting protein-protein interactions.
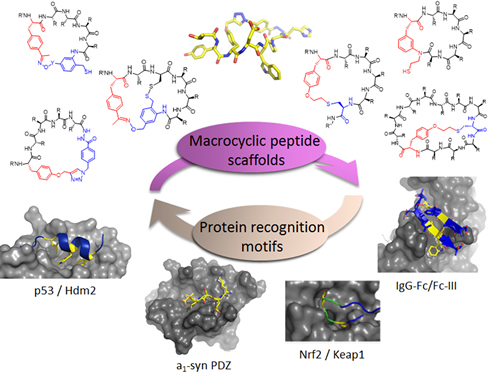
Figure 1. Diverse macrocyclic peptide scaffolds are being developed in the Fasan group for selective targeting of disease-related protein-protein interactions.
Our group has pioneered and introduced novel strategies for the modular construction of structurally diverse macrocyclic peptide scaffolds starting from genetically encoded polypeptide precursors. By integrating synthetic and biosynthetic tools, these methodologies have enabled the generation of ‘Macrocyclic Organo-Peptide Hybrids’ (MOrPHs), in which ribosomally produced peptide sequences are cyclized by means of non-peptidic linkers or genetically encoded unnatural amino acids. These strategies couple the versatility of chemical synthesis with the advantages of genetic encoding and power of genetic mutagenesis, providing unique opportunities for the creation and exploration of structurally diverse macrocycle libraries for the discovery of macrocyclic peptides capable of targeting different types of protein-protein interactions with high potency and selectivity (Figure 1). In ongoing projects, these methodologies are being applied to develop potent and selective inhibitors of protein-protein interactions (e.g., p53-Hdm2, Hedgehog/Patched) implicated in cancer development and progression (Figure 2). An integral component of these studies is the application of a variety of biophysical techniques (e.g. Surface Plasmon Resonance, NMR, fluorescence spectroscopy, X-ray crystallography) and biological assays for the investigation and characterization of the structural properties and biological activity of these compounds.
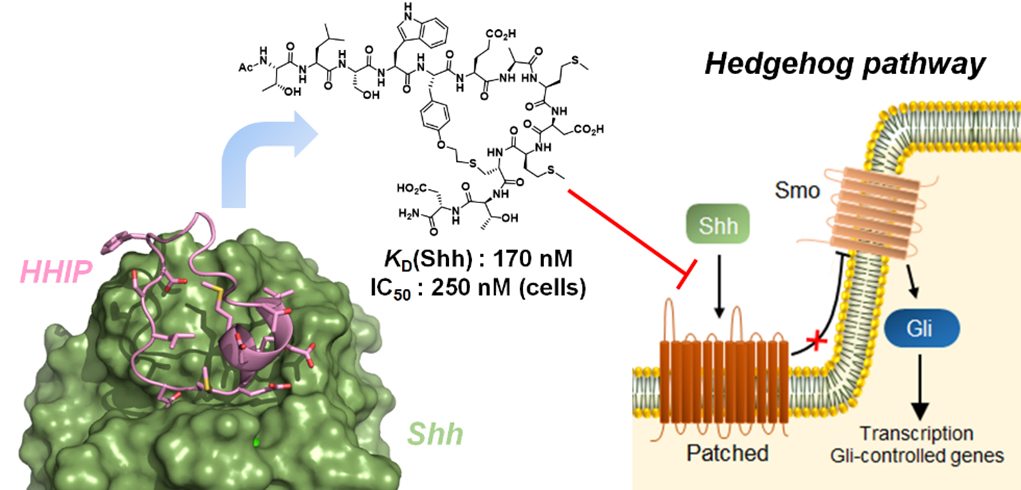
Figure 2. Macrocyclic peptide inhibitor of the Hedgehog signaling pathway.
Late-Stage C—H Functionalization and Diversification of Complex Molecules
Another major research area in the Fasan lab is concerned with the development and application of chemoenzymatic strategies for the selective, late-stage functionalization of aliphatic C—H bonds in complex organic molecules. Methods for the selective functionalization of unactivated C(sp3)—H bonds can streamline the synthesis of or but this represents one of the most challenging transformations in organic chemistry. Our group has been exploring the potential of engineered cytochrome P450s and P450-mediated chemoenzymatic synthesis as a way to address this challenge. A key goal in this area is the development of systematic strategies to modulate, predict, and ultimately, fine-tune the selectivity of P450 enzymes. Toward this end, we have introduced 'P450 fingerprinting' tools for rapidly mapping the active site configuration of these enzymes and predicting their reactivity toward various target substrates (Figure 3).
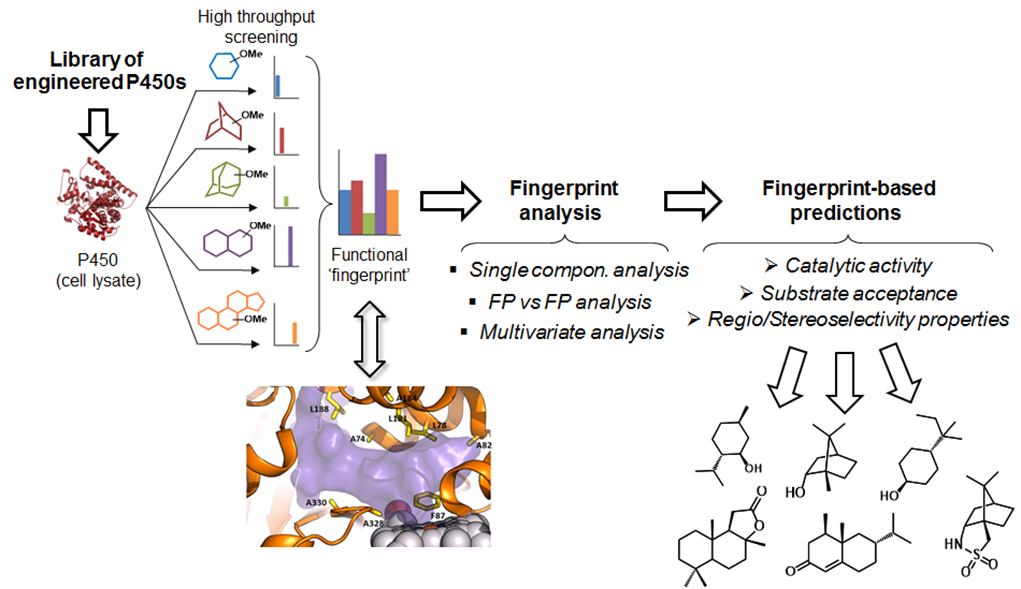
Figure 3. P450 fingerprint-based strategies for predicting P450 reactivity and guiding the development of site-selective P450 oxidation catalysts.
In ongoing projects, these methodologies are being applied to enable the late-stage functionalization of biologically active natural products and streamline the discovery of derivatives with improved pharmacological properties. For example, our group developed highly regio- and stereoselective P450 catalysts for the functionalization of multiple and previously inaccessible C‒H bonds in artemisinin, a key drug against multidrug-resistant malaria. In another project, we have leveraging P450-mediated chemoenzymatic synthesis for generating novel analogs of the antileukemic natural product parthenolide (called ‘parthenologs’) featuring improved activity and selectivity against leukemia stem cells and other types of cancers (Figure 4). Semisynthetic analogs of these natural products are also being applied to gain insights into the mechanism of action of these molecules and unveil potentially valuable new targets for cancer therapy.
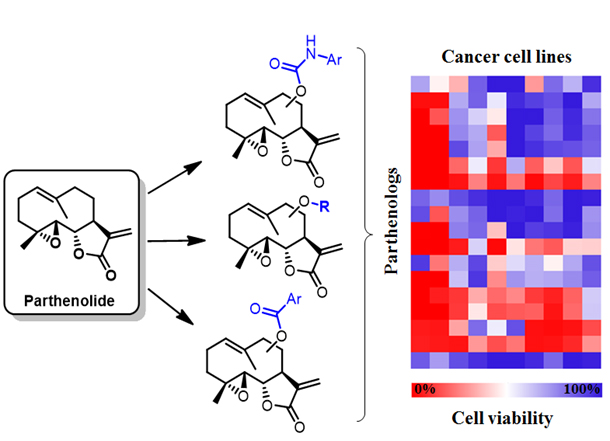
Figure 4. Parthenolide analogs with improved antileukemic activity and diverse anticancer activity profiles are generated via chemoenzymatic C‒H functionalization.
Engineered and Artificial Metalloprotein Catalysts for Biological Reactions
Enzymes can represent valuable catalysts for promoting chemical transformations with high degree of chemo-, regio-, and stereoselectivity and under mild reaction conditions. Unfortunately, many transformations useful for the synthesis of pharmaceuticals and other high-value compounds are not currently possible using natural enzymes. To address this gap, our group is designing and engineering metalloprotein catalysts capable of promoting a broad range of synthetically valuable carbon-carbon and carbon-heteroatom bond forming reactions which are not found in nature. Ongoing efforts have focused on developing biocatalysts based on heme-containing proteins (e.g., myoglobins and P450s) and other metalloprotein scaffolds for promoting nitrene- and carbene-mediated reactions (Figure 5). For example, our group has engineered P450 enzymes for catalyzing nitrene C−H insertion reactions, which are highly valuable transformations useful for the direct conversion of C(sp3)‒H bonds into C‒N bonds.
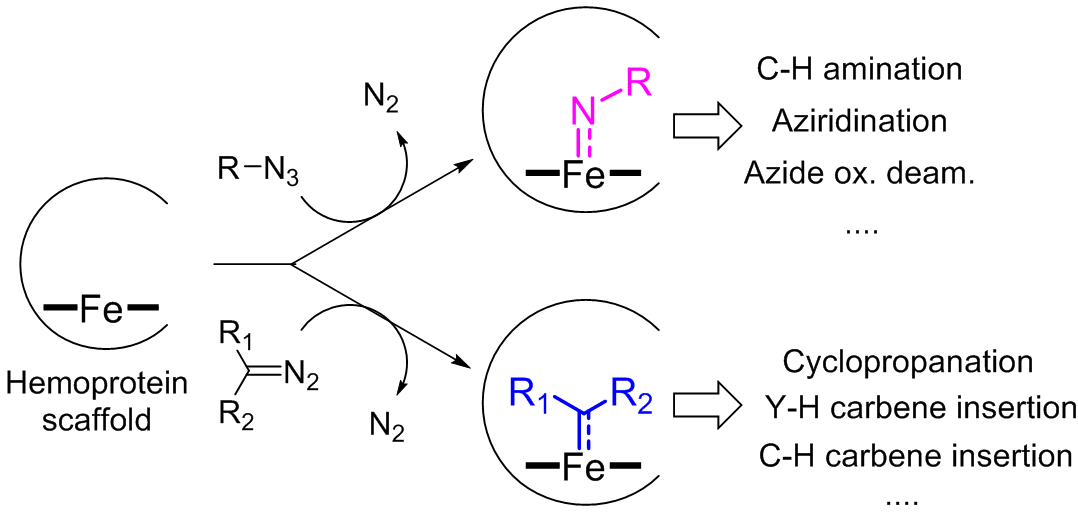
Figure 5. Representative biocatalytic nitrene- and carbene-mediated reactions developed in the Fasan group
In other projects, we have developed efficient myoglobin-based catalysts for promoting stereoselective olefin cyclopropanation reactions and these biocatalysts have been applied to the asymmetric synthesis of cyclopropane-containing drugs. Other ongoing projects within this area aim at tuning and enhancing the reactivity of these biocatalysts through the incorporation of non-native organometallic cofactors, also in combination with reengineering of the protein scaffold. All these efforts are complemented by mechanistic studies, using a combination of physical organic, computational, spectroscopic, and biophysical tools. These mechanistic insights gained are leveraged to guide the design and development of improved biocatalysts for current and new, abiological transformations.
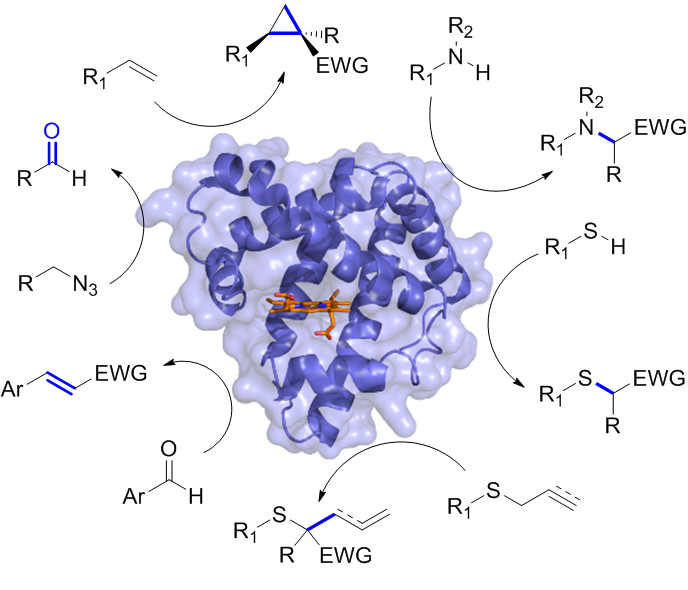
Figure 6. Non-natural reactions catalyzed by engineered myoglobin catalysts developed in the Fasan group.